SPANISH STUDY GROUP ON BRADYKININ-INDUCED ANGIOEDEMA (SGBA)
The SGBA was created in January 2007. It is a working group of the Spanish Society of Allergology and Clinical Immunology. Its coordinator is Dr. Teresa Caballero Molina. In 2011, it published the Consensus on the Epidemiology, Genetics, Clinical Symptoms, Diagnosis and Treatment of Angioedema Mediated by Bradykinin. At present (December 2015), the SGBA is composed of 19 allergists from Barcelona (2 Hospitals), Jaén, Logroño, Madrid (3 Hospitals), Seville (2 Hospitals), Talavera de la Reina, Valencia and Vigo
- Hospital Universitario La Paz de Madrid: Drs. Teresa Caballero (Coordinadora) Rosario Cabañas, Carmen Gómez Traseira and María Pedrosa
- Hospital Universitario Gregorio Marañón de Madrid: Drs. Mª Luisa Baeza and Alicia Prieto
- Hospital Universitario Severo Ocho de Legánes (Madrid): Dr. Nieves Prior
- Hospital Universitario Virgen del Rocío de Seville: Drs. Teresa González-Quevedo and Macarena Piñero (ex-Residente).
- Hospital Universitario Vall d’Hebron de Barcelona: Drs. Mar Guilarte and Anna Sala
- Hospital de San Pedro de Logroño: Dr. Teófilo Lobera
- Complejo Hospitalario de Jaén: Dr. Blanca Sáenz de San Pedro
- Complejo Hospitalario Universitario de Vigo: Dr. Carmen Marcos
- Hospital Universitario y Politécnico La Fe de Valencia: Ramón Almero Ves and Dr. Angel Campos (Retired)
- Hospital Nuestra Sra. del Prado de Talavera de la Reina (Toledo): Dr. Jesús Jurado
- Hospital Universitario de Bellvitge de Barcelona: Dr. Ramón LLeonart.
- Hospital Universitario Virgen Macarena de Sevilla: Dr. Amparo Conde
- M. Concepción López-Serrano worked for more than 40 years in the Hospital Universitario La Paz and was a member of the GEAB from the time it was founded until March 2015. Since retiring in 2013, she has been actively collaborating with AEDAF
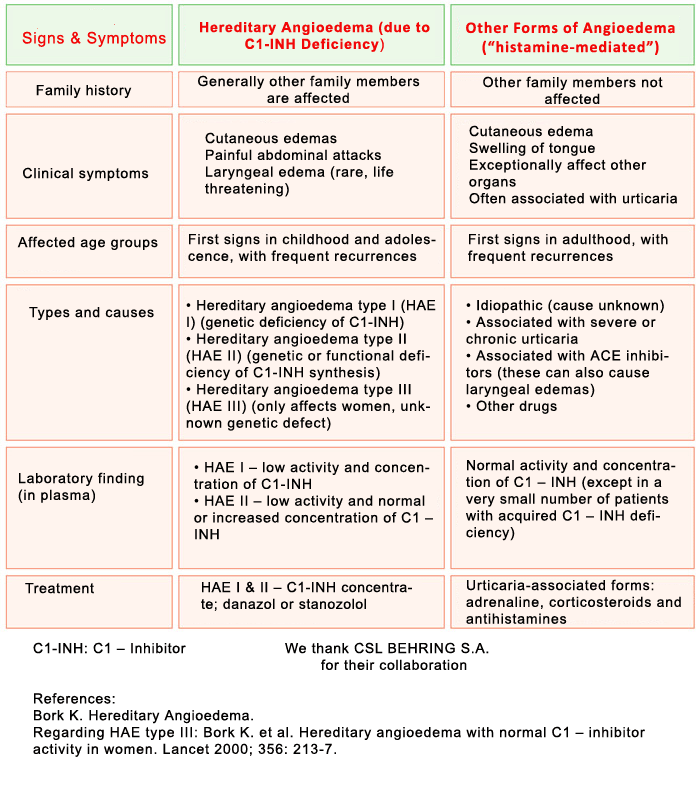
What is Angioedema from C1-Inhibitor Deficiency?
DEFINITION AND CLASSIFICATION
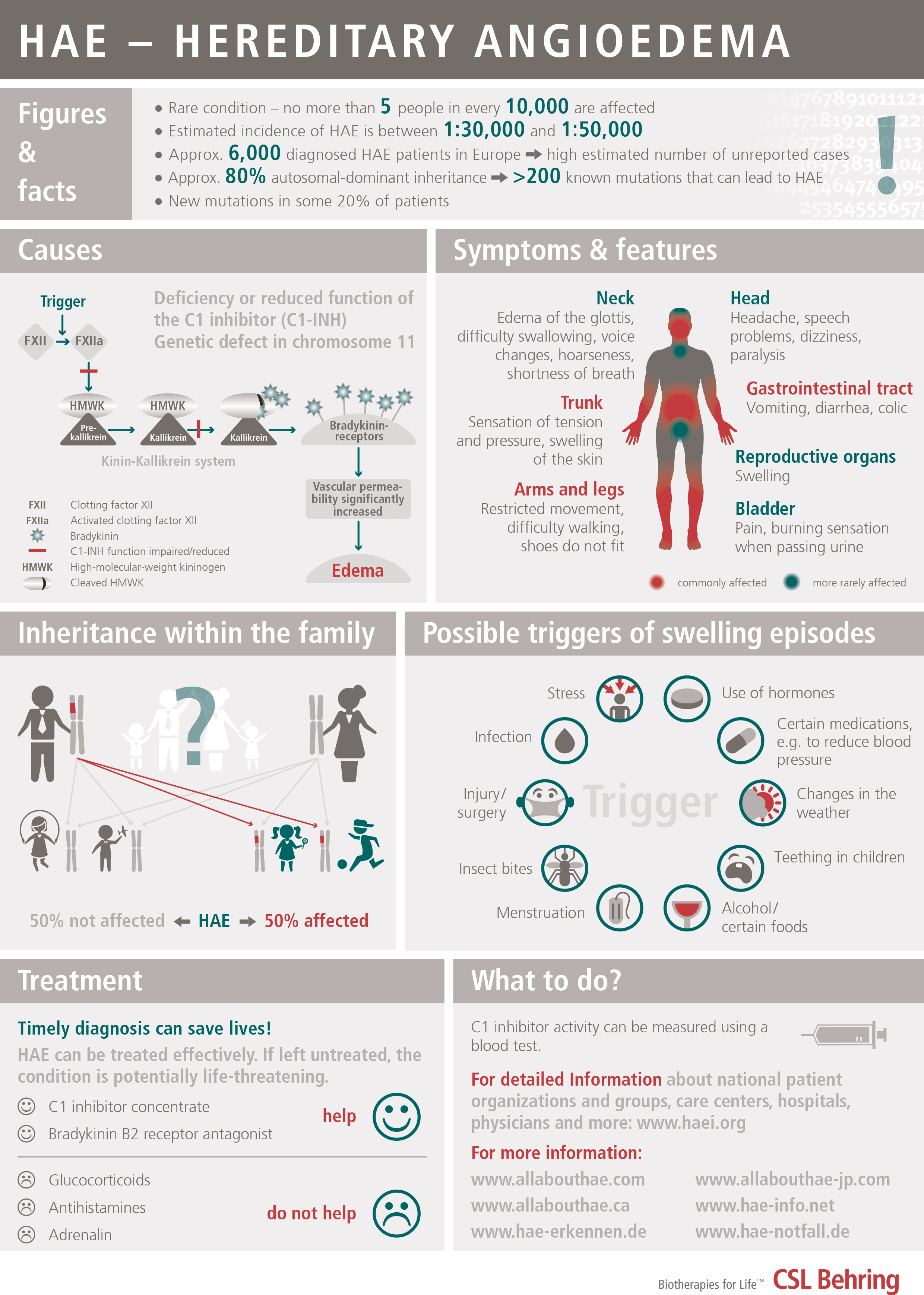
Variant of angioedema, due to a deficiency of the inhibitor of the first component of the Complement System, C1-Inhibitor, which can be caused by a genetic defect (hereditary angioedema or HAE) or by excessive consumption (acquired angioedema). It is estimated that the hereditary form may affect 1 in every 50,000 people. It is transmitted on a dominant autosomal basis (i.e., each child has a 50% chance of inheriting it if one of the two parents has it). In up to 25% of cases, there is a spontaneous mutation (the parents do not have it, the patient is the first to have the genetic defect and from then on it can be inherited by his/her children).
There are few cases of the acquired form, less than 300 in the world.
There is a new form of hereditary angioedema with normal C1 inhibitor levels that primarily affects women and is related to high levels of exogenous estrogens (oral hormonal birth control pills, hormone replacement therapy, etc.) or endogenous estrogens (pregnancy). It is called hereditary angioedema without C1-esterase inhibitor deficiency. A mutation of coagulation factor XII has been described in a subgroup of patients as the cause of this disorder (HAE-FXII).
DISEASE SYMPTOMATOLOGY
C1 inhibitor deficiency may be asymptomatic.
The disorder is usually characterized by swelling lasting from 2 to 5 days that recurs relatively frequently on a variable basis, and that affects both extremities and internal organs; there is an onset period of progressively increasing intensity of 6 to 24 hours, and it spontaneously subsides or resolves in 12 to 36 hours (without applying treatment). It is not associated with urticaria. The swelling is white or exhibits a pinkish rash, with compacting of the affected area until it gets very hard and mobility is lost in the area and a period of stability when the skin has a whitish coloring. On occasions it is accompanied by itching or a tingling sensation.
It can affect:
- Subcutaneous tissue: face, neck, shoulders, extremities (hands, feet, arms, legs), buttocks, genitals
- Submucosal tissue of abdominal organs: stomach, intestine, bladder
- Submucosal tissue of the upper respiratory tract: tongue, throat, pharynx and larynx
Swelling of the digestive tract causes abdominal pain and vomiting and it can simulate an acute appendicitis. Swelling of the respiratory tract can cause edema of the glottis with hoarseness and voice loss and in some cases asphyxia.
PRECIPITATING FACTORS
Traumatisms; Some forms of intense physical exercise (e.g., cycling); Surgical operations: Dental extractions; Fatigue; Insomnia; Stress; Disappointment and distress; Infections; Menstruation; Estrogens (oral contraceptives, hormone replacement therapy); antihypertensive drugs of the ACE (angiotensin converting enzyme) inhibitor group (ACEi).
DIAGNOSIS
Since this disorder may be similar in its presentation to other forms of angioedema caused by allergies or other medical conditions, a correct diagnosis of HAE is very important to avoid potentially fatal consequences such as asphyxia or unnecessary abdominal surgery.
As a screening technique, levels of C4 are measured, which are lower than normal values both during the crisis period (i.e., swelling) and during periods without symptoms. If these levels are low or even normal, the levels of C1-inhibitor and the functional activity of C1-inhibitor must be determined. Low serum levels of C1-inhibitor or of its functional activity confirm the diagnosis.
In the case of hereditary angioedema without C1-esterase inhibitor deficiency, the antigenic and functional C4 and C1 inhibitor levels are usually normal.
TREATMENT
Severe attacks (upper respiratory, abdominal and moderate-severe cutaneous swellings): Intravenous C1-INHIBITOR plasma concentrate (Berinert® 20 U/kg or Cinryze® 1000U), or icatibant acetate (Firazyr®), 30 mg subcutaneous. If the attack does not subside shortly depending on the criticality of the affected area, administer an extra dose of Berinert® or Cinryze® one hour later (the dose of Firazyr® cannot be repeated until at least 6 hours pass). It is advisable to begin treatment as early as possible. If these products are NOT available, intravenous tranexamic acid or fresh frozen plasma may be used although they are much less effective.
Long-term prevention: Attenuated androgens (DANAZOL – Danatrol®) or STANOZOLOL (Winstrol®), or tranexamic acid. The dose is personal, indicated by the doctor. In serious cases that do not respond to attenuated androgens or tranexamic acid, or in the event of significant side effects caused by these drugs or of underlying diseases in which they are contraindicated, intravenous C1-INHIBITOR plasma concentrate may be used on a regular basis. (Cinryze® 1000U two times a week and adjust the interval depending on response.)
Short-term prevention (surgical operations, dental manipulation, or oropharyngeal procedures): Increase the dose of Danazol/Stanozolol from 6 days before until 3 days after, or administer 1000 units of intravenous C1-Inhibitor (regardless of weight) 1-6 hours before operation (as soon as possible before operation). The treatment of choice will be C1-Inhibidor plasma concentrate. If C1-Inhibitor is NOT available, fresh frozen plasma may be administered at least one hour before the operation.
The treatment of hereditary HAE without C1-esterase inhibitor deficiency (formerly Type III HAE) is more difficult, since there are still no drugs on the market. For severe attacks, there are isolated cases that have been treated with plasma derived C1-inhibitor concentrate or icatibant acetate.
Other recommendations:
- avoid estrogens (contraceptives
- avoid antihypertensive drugs inhibiting the ACE: angiotensin converting enzyme
- vaccination against Hepatitis A and B
VERY IMPORTANT: antihistamines, corticoids and adrenaline are not effective.
HAE SYMPTOMS